
Guantes, condones, jeringas y agujas. Esos son los únicos elementos de uso médico sometidos a control sanitario en el país. El resto, desde gasas, catéteres y bisturíes, pasando por bombas de insulina, implantes de cadera o marcapasos, hasta ventiladores artificiales o equipos de resonancia magnética, se pueden fabricar, importar y vender sin autorización. LaBot analizó la base de datos de la FDA: en Chile se venden algunos de los dispositivos que han reportado fallas.
“No veía nada, ni una luz… nada, nada”. A mediados 2013 Luis Marmolejo perdió la visión de su ojo derecho luego de ser sometido a una cirugía oftalmológica por desprendimiento de retina. El procedimiento se realizó en el Hospital Regional de Rancagua, donde le aplicaron perfluootoctano, un dispositivo médico fabricado por la empresa turca Meran Tip.
“Yo me fui a operar y pasó el tiempo y no veía nada. El médico después me dijo que ya no tenía remedio, que iba a perder el ojo”, señaló Marmolejo en una entrevista con Cooperativa en agosto de 2013.
Según el Instituto de Salud Pública (ISP), hubo siete notificaciones de “eventos adversos”, es decir, de “daño no intencionado al paciente, usuario u otra persona, que ocurre como consecuencia del uso de un dispositivo médico”, relacionados con el plerfuootoctano. En este caso, el daño fue la “pérdida de la visión del ojo intervenido”. Y la medida adoptada, el retiro voluntario del dispositivo del mercado.
La empresa Meran Tip vendía el producto en el país desde 2012. Al momento de la crisis, se había distribuido a seis hospitales públicos y seis centros de salud privados. El organismo lanzó una alerta sanitaria y la empresa retiró el producto del mercado. Y aunque el perfluooroctano estaba calificado como un producto de “alto riesgo”, el propio ISP reconoció oficialmente que no podía regular su venta: “Este producto actualmente no se encuentra en control sanitario”.
Cinco años después, los pacientes siguen tan desprotegidos como entonces: salvo cinco excepciones, Chile carece de una regulación para evaluar la calidad de más de 200 mil dispositivos médicos antes de autorizar su uso y comercialización. Y la definición es amplia: incluye elementos básicos como apósitos y catéteres; instrumental quirúrgico como bisturíes; elementos como bombas de insulina o lentes de contacto; implantes como prótesis de cadera, stents o marcapasos; máquinas como carros de resucitación o ventiladores artificiales; equipos como tomógrafos, y mobiliario médico.
Crédito: OjoPublico/SaludconLupa
Tampoco existe en Chile un sistema de alerta obligatorio que recopile las fallas de los dispositivos e informe a los pacientes o a la comunidad médica.
Así, mientras el objetivo de los dispositivos médicos es mejorar o salvar la vida de los pacientes, en Chile no hay un organismo que vele por su calidad. La extendida idea de que médicos y pacientes pueden confiar en que la autoridad ha dado un visto bueno a los dispositivos médicos es una fantasía.
Hace apenas nueves meses, en febrero de 2018, Tatiana Tobar, de la Subsecretaría de Salud, habló derechamente de “protección y resguardo insuficiente para los pacientes” en una “reunión técnica” que el Ministerio de Salud, el ISP y la Dirección de Relaciones Económicas de la Cancillería (Direcon) sostuvieron con representantes de la industria de los dispositivos médicos.
En el encuentro Tobar señaló que en Chile existe un “retraso regulatorio”, “dificultad para fiscalizar (inhibición)” y un “lento desarrollo de la tecnovigilancia”, es decir, del comportamiento de los dispositivos médicos una vez que están en uso.
FALLA 1: ESCASA REGULACIÓN
Recién en 1997, a través de una modificación al Código Sanitario, Chile definió legalmente qué son los dispositivos médicos. En 1998, el Ministerio de Salud dictó el “Reglamento de control de productos y elementos de uso médico”. En su artículo 22 definió que mediante decretos dictados por esa cartera “se hará efectiva progresivamente la aplicación de este reglamento a los distintos dispositivos y elementos médicos regulados por él”.
Veinte años después, se han dictado sólo dos decretos y la regulación aplica sólo a cinco dispositivos médicos: guantes quirúrgicos de caucho, guantes de caucho para exámenes médicos y condones de látex de caucho, todos desde 2004; y agujas y jeringas hipodérmicas para un solo uso, desde 2007.
Aunque el reglamento de 1998 creó cuatro tipos de dispositivos médicos, desde los que “presentan un grado muy bajo de riesgo” (Clase 1) hasta los “los más críticos en materia de riesgos” (Clase 4), en Chile ningún dispositivo médico perteneciente a esta última categoría –entre ellos, los implantes– está sometido a control sanitario.
Si una persona sufre, por ejemplo, un infarto, y alcanza a ser atendida en una ambulancia o en un centro de salud, probablemente será socorrida utilizando un carro para resucitación. Y, en caso de sobrevivir, quizás la sometan a una angioplastía y le implanten un stent, es decir, una malla para abrir la arteria. Ni el equipo ni el implante, por ejemplo, están sometidos a control sanitario. Tampoco la mayoría de los implementos utilizados en la atención de emergencia o en pabellón.
–Una empresa fabrica o importa un marcapasos, ¿puede venderlo sin pedir autorización?
–Lamentablemente sí —responde Stephan Jarpa, ex director del ISP entre 2013 y 2014, hoy director de la agencia InHouse, que asesora a la industria de los dispositivos médicos.
La calidad de los dispositivos médicos que se venden en Chile depende, entonces, de la voluntad de dos actores. Por un lado, de las empresas que los fabrican, importan (el 95,5% del mercado, según el ISP) o comercializan. Y, por otro, de los servicios de salud públicos y privados que los compran, incluyendo a los médicos que los solicitan.
Hay empresas en Chile que sólo importan dispositivos certificados en el extranjero y centros de salud que sólo compran este tipo de dispositivos. Pero también hay empresas y centros de salud que no se autoimponen estos estándares. A esto se suma, como muestra la investigación del Consorcio Internacional de Periodistas de Investigación(ICIJ, por su sigla en inglés), que en aquellos países donde hay regulación es necesario mejorarla, pues muchos implantes no están siendo sometidos a un debido control sanitario antes de ser comercializados.
En Chile, las autoridades, las empresas y los médicos saben que en algunos casos –no pocos– se venden y utilizan dispositivos médicos de baja calidad. Y que no siempre los pacientes están protegidos.
Según el ex director del ISP, Stephan Jarpa, en Chile entre el 30% y 40% del mercado de los dispositivos médicos cumple la norma internacional ISO 13485, el estándar mundial actual. “En Chile no se la están exigiendo a nadie y más del 50% no la tiene”, dice. “Si se exigiera, lo más probable es que varios proveedores saldrían del mercado”, agrega.
De hecho, según la Organización Mundial para la Salud, Chile está entre los países de América Latina con peor regulación para dispositivos médicos, detrás de Argentina, Brasil, Colombia y Uruguay –que cumplen el estándar más alto–, y de Bolivia, Perú, Paraguay y Ecuador. Solamente Venezuela, está peor que el país en esta materia.
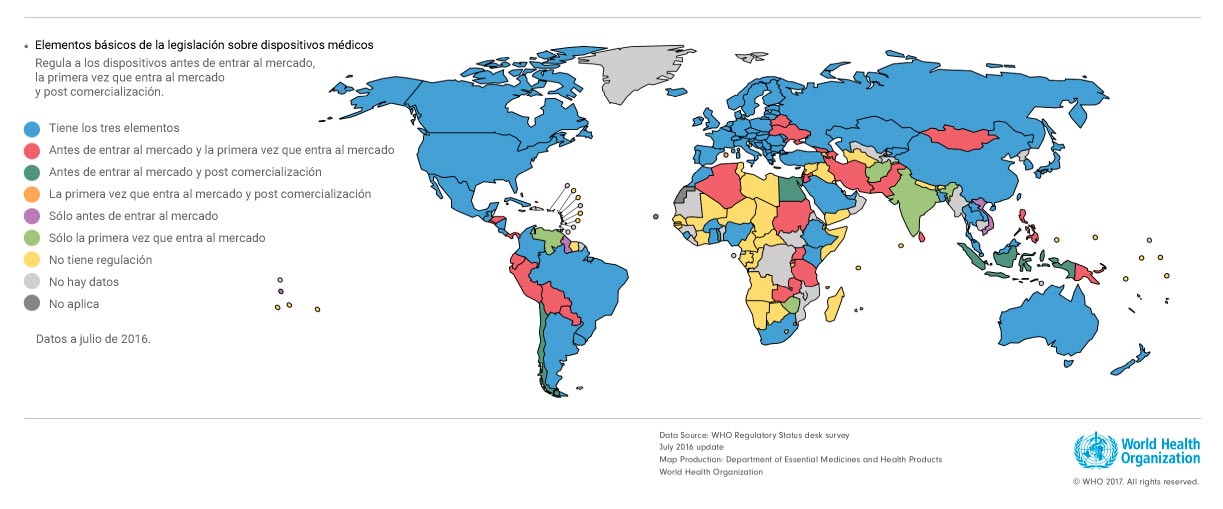
Así y todo, no ha habido voluntad de regular este mercado.
Peor aún: hay opacidad. Según esta investigación de LaBot, y pese a la escasez de información oficial, hay datos consistentes que muestran lo obvio: en un país sin regulación, hay dispositivos que fallan.
FALLA 2: NO HAY RASTRO
LaBot solicitó en agosto pasado, vía Ley de Transparencia, “copia de todas las notificaciones sobre eventos o incidentes adversos ocasionados por dispositivos médicos, así como las medidas y resoluciones gatilladas por dichas notificaciones” desde 2008 en adelante. El ISP respondió con un “Resumen de las notificaciones más relevantes recibidas por el Sistema de Tecnovigilancia”.
Según el Instituto de Salud Pública, en los últimos diez años sólo dos productos sometidos al control sanitario arrojaron problemas:
- En 2013 y 2014, las jeringas Luer Luck de 10 cc con agujas fueron reportadas por generar “incidentes adversos”, es decir, “potencial riesgo de daño no intencionado al paciente o usuario u otra persona”, pero que fue evitado y “no generó un desenlace adverso”.
- En 2016, los preservativos Kaiju, fueron reportados por “defectos de calidad”.
En ambos casos los fabricantes eran empresas chinas. En el primero, según señala la respuesta obtenida vía Ley de Transparencia, la empresa retiró voluntariamente las jeringas. En el segundo, el ISP ordenó sacar los condones del mercado.
En el caso de los dispositivos que no están sometidos a control sanitario, el ISP también informó sobre dos casos:
- En 2010, el ISP se hizo eco de una alerta sanitaria internacional para retirar y reemplazar los implantes mamarios franceses Poly Implant.
- En 2013, el ISP recibió las notificaciones de “eventos adversos” por el perfluootoctano, el mismo que provocó la pérdida de visión del ojo derecho de Luis Marmolejo.
Según el ISP, “las notificaciones recibidas son mayoritariamente de defectos de calidad de los distintos dispositivos médicos y no han llevado a un evento adverso a los pacientes o usuarios”.
¿Es posible que en la última década sólo cuatro dispositivos médicos hayan generado problema entre los pacientes chilenos? ¿Es posible algo así en un país donde no son sometidos a control de calidad y donde se comercializan alrededor de 200 mil dispositivos médicos? ¿Y es posible que sólo un dispositivo implantable haya generado un “evento adverso”? La más reciente investigación de ICIJ da cuenta de que eso es prácticamente imposible.
La regulación en Chile no sólo es casi inexistente para autorizar el uso y la venta de dispositivos médicos en el país. Tampoco existe un macizo sistema de monitoreo post comercialización.
Desde hace algunos años, el ISP ha hecho un esfuerzo por hacer un seguimiento del comportamiento de los dispositivos médicos. A través del Departamento de Tecnovigilancia, el organismo recibe notificaciones de profesionales de la salud en centros públicos y privados, y de las empresas importadoras o fabricantes. En el caso de los dispositivos médicos sometidos a control sanitario –los guantes, condones, agujas y jeringas– las notificaciones son obligatorias por ley. En el resto, son voluntarias.
Con estos datos, el ISP publicó el “Boletín de Tecnovigilancia: Estadísticas 2015-2017” sobre “incidentes adversos” o “eventos adversos” provocados por dispositivos médicos en ese periodo.
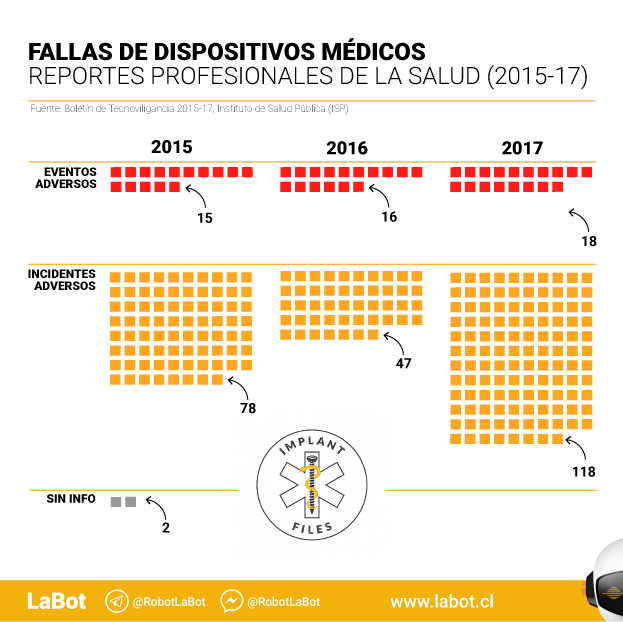
En total, hay 431 reportes. De ellos, 91 corresponden a “eventos adversos”, es decir, aquellos que implican algún daño a pacientes. Un promedio de 30 casos por año.
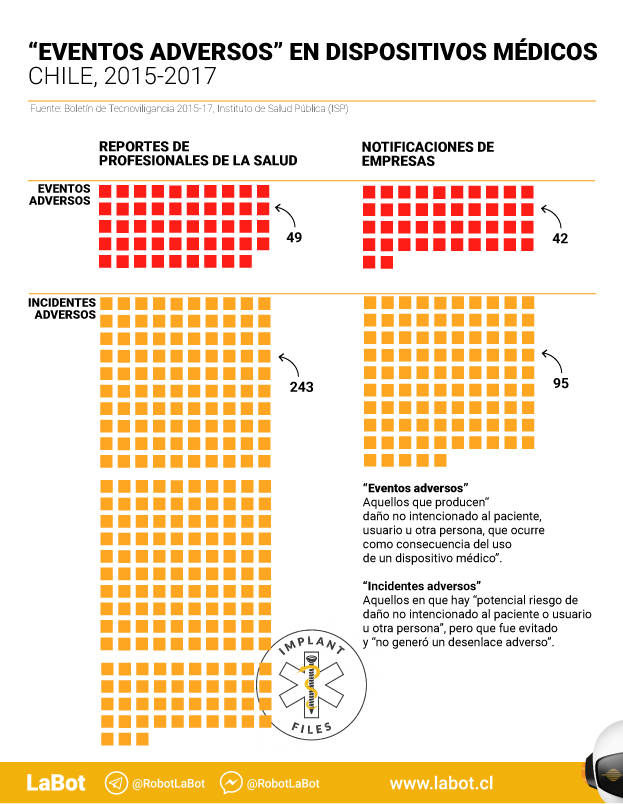
Y sabemos que esa cifra es incompleta y parcial. El boletín, de 14 páginas, señala explícitamente que el registro es incompleto “no correspondiendo a un sistema de notificación de carácter obligatorio o en red, siendo susceptible de una probable subnotificación por parte de usuarios (profesionales de la salud) y empresas.”
OPACIDAD
El “Boletín de tecnoviligancia: Estadísticas 2015-2017” distingue los reportes de profesionales de la salud de los de las empresas.
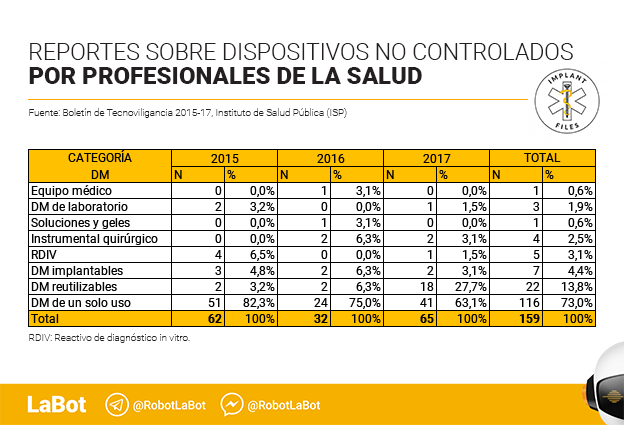
Las notificaciones realizadas por profesionales de la salud llegan a 294 y representan un 68,2% del total. En este caso, más de la mitad de las notificaciones (159) se refieren a 56 dispositivos médicos sin control sanitario.
En esta sección hay notificaciones sobre 49 eventos adversos, es decir, aquellos que pueden dañar la salud. Pero no hay información sobre si se trata de dispositivos sometidos (o no) a control sanitario. Ni sobre qué tipo de productos –elementos de uso médico, implantes, medios de contraste, software o máquinas, por ejemplo– fallaron.
El informe señala, eso sí, que de las 159 notificaciones sobre dispositivos médicos no controlados “73% (116/159) correspondió a dispositivos médicos de un solo uso, principalmente apósitos (18) y catéter intravenoso (14); seguido de dispositivos reutilizables con el 13,8% (22/159), con 13 notificaciones correspondientes a aerocámaras; (y) 4,4% (7/159) a dispositivos implantables”.
(Foto: Robina Weermeijer on Unsplash)
¿Cuáles son los implantes que generaron eventos adversos en los pacientes? ¿Qué empresas los fabricaron? ¿Qué fallas arrojaron? ¿Hubo algún paciente que sufriera deterioro en su salud o daño?
El 5 de noviembre, LaBot solicitó una entrevista formal con Janepsy Díaz, jefa del Departamento de Dispositivos Médicos del ISP, creado en mayo de 2017. Casi diez días después, el ISP informó que Díaz no podría otorgar la entrevista por “motivos de agenda”. LaBot reiteró la petición, amplió los plazos y solicitó otra entrevista con la directora (S) del ISP, Judith Mora. El viernes 16 envió dos cuestionarios pidiendo respuestas por escrito. El 20 de noviembre el ISP insistió en que no daría entrevistas.
LO QUE INFORMAN LAS EMPRESAS
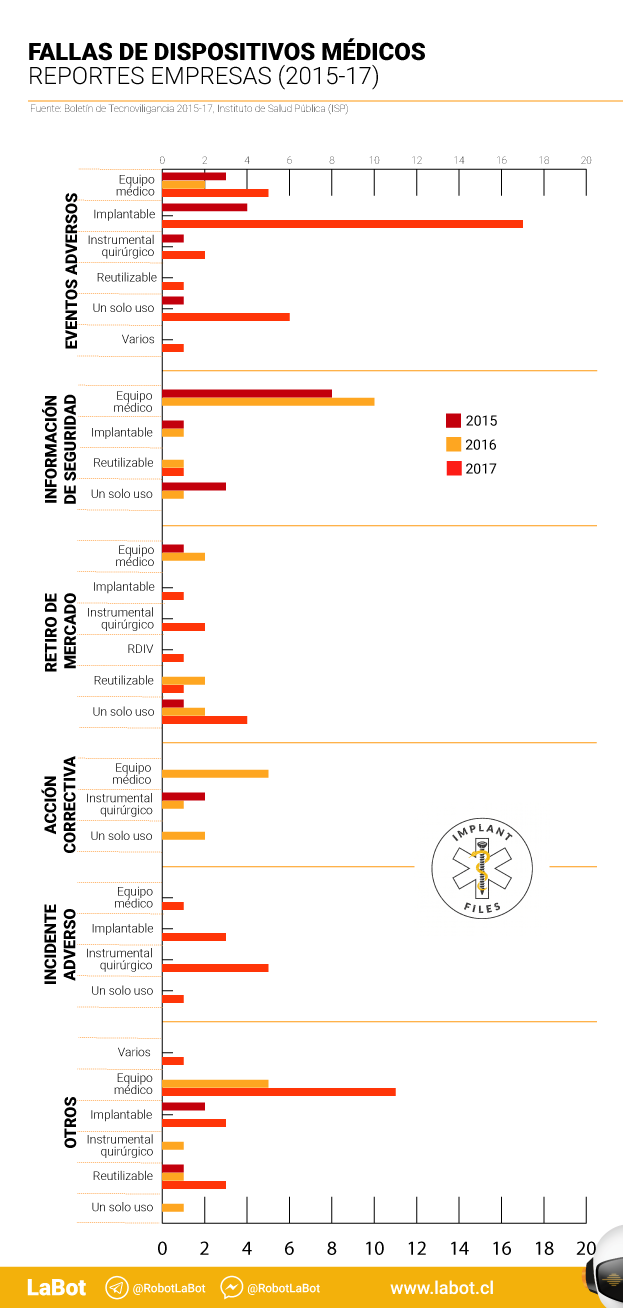
La otra sección del “Boletín de Tecnovigilancia: Estadísticas 2015-2017” recopila las notificaciones realizadas por las empresas fabricantes, importadoras y/o distribuidoras de los productos. En este caso los reportes llegan a 137, pero el ISP no distingue si alguno de estos dispositivos está sometido a control sanitario. En las notificaciones hechas por las empresas, 32 afectan a implantes, 21 de ellas con “eventos adversos”. 2017 tuvo un peak con 17 de estos últimos casos. Al comparar las dos secciones del informe, las empresas reportan más fallas de dispositivos implantables que los profesionales de la salud (32 versus 7). Otro caso en que las notificaciones desde las empresas son mayores que las hechas por profesionales de la salud ocurre en la categoría equipos médicos: 52 reportes versus 1. En las notificaciones de las empresas, de hecho, nueve corresponden a casos de “eventos adversos”.
(Foto: Piron Guillaume on Unsplash)
¿A qué dispositivos corresponden? ¿Hay productos que fallan más que otros? ¿Quiénes son los fabricantes? De nuevo, no tenemos cómo saberlo. A pesar de que en el caso de los medicamentos el ISP posee un “Sistema de consulta de Productos Registrados” en su página web, en la sección tecnovigilancia la información es escasa y parcial. LaBotpreguntó al ISP por qué no existe un registro público sobre dispositivos médicos, pero, como se explicó antes, el organismo no respondió. La investigación realizada por ICIJ entrega una idea sobre la utilidad de hacer pública esta información: permite a pacientes y médicos explorar avisos de retiro, alertas y advertencias de seguridad de fabricantes. La Base de datos internacional de dispositivos médicos (IMDD, por sus siglas en inglés, contiene 70 mil registros sobre 11 países y se seguirá actualizando.
LOS RECALLS DE LA FDA
Hasta el 20 de junio de 2018, en Chile había al menos 463 empresas fabricantes, importadoras y/o distribuidoras de dispositivos médicos inscritas voluntariamente en el ISP (algunas de ellas son también exportadoras). Si el registro fuese obligatorio, el número total sería mayor.
En el “Boletín de Tecnovigilancia: Estadísticas 2015-2017”, sin embargo, no existe información sobre cuáles son las empresas que fabricaron los dispositivos médicos que han sido reportados por provocar “eventos adversos”, es decir, que pueden haber dañado a los pacientes.
El dato que el informe sí entrega es el lugar de origen de los productos. En el caso de los reportes realizados por profesionales de la salud, que incluyen “eventos” e “incidentes adversos”, el ranking lo encabeza Asia con 131 notificaciones, seguido por Chile, con 45. Europa está en el tercer lugar, con 19 reportes. Norteamérica aparece recién en el cuarto lugar, junto al resto de Sudamérica, con 16 notificaciones en cada caso.
El ranking de notificaciones de las empresas es distinto. Lo encabeza Norteamérica –específicamente Estados Unidos– con 91 notificaciones. Y hay 22 reportes sobre productos que provienen de Asia, Centroamérica, Sudamérica y Europa, en conjunto. Para el resto de los casos –24– no hay información.
Como el ISP no informa qué dispositivos médicos arrojan fallas ni cuáles son las empresas que los fabrican, LaBottrabajó con la base de datos de la FDA. El organismo estatal que fiscaliza los dispositivos médicos en Estados Unidos –y cuyas debilidades han quedado expuestas en la investigación de ICIJ–, posee un registro público con los productos que han sido corregidos o retirados del mercado porque han provocado daño, problemas de salud o la muerte, o han presentado fallas en su funcionamiento que los vuelve potencialmente peligrosos.
LaBot analizó la base de datos de la FDA entre junio de 2012 y abril de 2018. En ese período se realizaron al menos 860 recalls o avisos de retiro del mercado para algo más de 2.730 productos que, según la FDA, fueron comercializados en Chile.
La FDA clasifica estos recalls o alertas sobre retiro de productos en tres categorías: Clase I, para los dispositivos que pueden provocar serios problemas de salud o la muerte; Clase II, para los que pueden afectar la salud de manera temporal o reversible; Clase III, para los casos en que las fallas de los dispositivos no dañan ni afectan la salud.
Según la base de datos analizada, en el caso de Chile la FDA registra al menos 75 recalls Clase I –los más graves–que afectan a poco más de 170 dispositivos médicos. En promedio, más de 28 productos por año. Luego aparecen 760 recalls Clase II que afectan a más de 2570 productos distintos. Por último, arroja 29 recalls Clase III para casi 40 dispositivos.
Entre los dispositivos con recalls Clase I hay catéteres y tubos traqueales, bombas de insulina, marcapasos, implantes para estimulación eléctrica, respiradores artificiales, incubadoras y máquinas para realizar tomografías. Y entre las empresas con 10 recalls o más aparecen Teleflex Medical (Arrow International), Vyaire (Carefusion), Medtronic (Covidien), GE Healthcare, Cook, Biomeriuex y Nova Biomedical Corporation.
LaBot buscó los más de 170 dispositivos médicos que aparecen en los recalls Clase 1 de la FDA en la base de datos DataSur, una plataforma que contiene declaraciones de importaciones y exportaciones de diversos países. Ahí encontró 25 productos por coincidencia de nombre y modelo, o código (una lista que podría crecer, pues los datos no siempre están ingresados de la misma forma). LaBot no pudo confirmar, sin embargo, si se trata de los mismos lotes, pues esa información no aparece en DataSur.
Entre estos dispositivos médicos hay catéteres de distintos tipos, máquinas para administrar anestesia, respiradores artificiales, equipos de asistencia ventricular y un equipo de resonancia magnética. Hay también kits de prueba de plomo en la sangre. Y tiras para medir glucosa, así como kits para bombas de insulina. Aparecen además electrodos para implantes de estimulación cerebral, que se utilizan para tratar enfermedades como el parkinson, la distonía y la epilepsia.
LaBot buscó estos últimos productos en Mercado Público, la plataforma que usan los organismos públicos para comprar insumos. Encontró que al menos una docena de estos dispositivos, afectados por recalls de la FDA, han sido comprados por servicios de salud. Pero, de nuevo, no existe información pública para verificar en qué casos se trata de los mismos lotes.
Por último, según un análisis realizado por el medio peruano Ojo Público a partir de los datos de la FDA, entre 2009 y comienzos de 2018 desde Chile se realizaron 64 notificaciones de eventos adversos. En ninguno se reportó la muerte de algún paciente, en un caso sí se informó sobre dañor severos y en los restantes 63 se reportaron fallas en los dispositivos.
NUEVA REGULACIÓN
En noviembre de 2015, en el marco de la discusión de la primera versión de la Ley de Fármacos II, el entonces subsecretario de Salud, Jaime Burrows, anunció que el gobierno enviaría indicaciones en cinco áreas. Entre ellas “la regulación de los dispositivos médicos”.
“Es un área no regulada claramente en nuestra legislación. En el ISP se registran los dispositivos, que son los guantes quirúrgicos, los condones y materiales que tienen que ver con inyecciones, jeringas y agujas; pero otro tipo de implementos no se someten a un control sanitario que se necesita para ponernos a nivel internacional”, afirmó Burrows en esa ocasión.
Recién en enero de 2017, el gobierno de Michelle Bachelet presentó las indicaciones. El Senado aprobó el proyecto en enero de 2018 y pasó a la Cámara de Diputados, que todavía no ha iniciado su discusión.
En lo grueso, la iniciativa actualiza la definición de dispositivos médicos, crea un registro obligatorio de los “elementos de uso médico” que se fabrican o importan, y fija como requisito que los dispositivos considerados “de riesgo sanitario” cuenten con “registro sanitario”.
Además, los fabricantes, importadores y distribuidores deberán inscribirse en el ISP antes de iniciar sus actividades y, en algunos casos, contar con autorización sanitaria. Por último, se prohíbe explícitamente la “fabricación, importación, tenencia, distribución y transferencia” de dispositivos “falsificados, adulterados, alterados o contaminados”.
La mayoría de las indicaciones incluidas en la Ley de Fármacos II busca hacer obligatorios procedimientos que hoy son voluntarios.
En años recientes, por ejemplo, el ISP ha alentado a las empresas que comercializan dispositivos médicos a inscribirse voluntariamente en el registro del organismo. Además, desde el 2 de julio de este año, todos dispositivos médicos que ingresan al país deben obtener un Certificado de Destinación Aduanera (CDA), aunque no estén sometidos a control sanitario. Una medida que le permitirá al ISP tener, por primera vez, un registro completo de los dispositivos importados, y servirá para fiscalizar el ingreso de productos fraudulentos, los que según la OMS alcanzan el 8% a nivel mundial.
EL LOBBY DE LA INDUSTRIA
Dos meses después de asumir, el 7 de mayo de 2018, el gobierno de Sebastián Piñera retiró las últimas indicaciones enviadas por la administración anterior al proyecto de Ley de Fármacos II e ingresó las propias. En el caso de los dispositivos médicos, el texto aprobado por el Senado se mantuvo casi intacto. Visto así, pareciera que hay consenso.
Pero la industria, o al menos parte de ella, se ha movilizado en contra del proyecto.
En diciembre de 2015, un mes después de que el ex subsecretario Burrows anunciara que el Ministerio de Salud enviaría indicaciones para regular los dispositivos médicos en Chile, se formó la Asociación de Proveedores de la Industria de la Salud AG (APIS). Ad portas de que se retome la tramitación de la Ley de Fármacos II, el lobby se ha concentrado en frenar la regulación de los dispositivos médicos tal y como está propuesta.
Según la plataforma Ley Lobby, en agosto la directiva de APIS, encabezada por su presidente Cristián Hänel (Tecnigen) y su director ejecutivo Eduardo del Solar, se reunieron con la directora (S) del ISP, Judith Mora. Entonces manifestaron la “preocupación” de APIS por la implementación de la Ley de Fármacos II, en lo que se refiere a los dispositivos médicos. Según el registro de la audiencia, el ISP les comunicó que serían llamados a mesas de trabajo en las que el organismo presentará la normativa que ha trabajado con la asesoría del BID en la materia.
Dos meses después, en octubre de 2018, Del Solar y otros representantes de APIS sostuvieron otro encuentro, esta vez con la jefa del Departamento de Dispositivos Médicos del ISP, Janepsy Díaz. Según el acta de la reunión, APIS señaló que “está de acuerdo con la regulación de dispositivos médicos, pero no con la denominación (…) como ‘Elementos de uso médico’ ni con que se les considere un bien público”.
APIS no es la única asociación gremial que ha intercedido ante la autoridad. Desde 2017, distintos funcionarios del ISP han sostenido al menos nueve reuniones con gremios del rubro para tratar el tema.
Ha sido APIS, al menos públicamente, la que ha expresado una postura más dura. En una carta enviada el 24 de julio al presidente de la Comisión de Salud de la Cámara de Diputados, el socialista Juan Luis Castro, manifiestan “serios reparos” al proyecto. Advierten que, de mantenerse el plazo de 12 meses para inscribir cerca de 200 mil dispositivos médicos “sobrevendrá un colapso en el sistema hospitalario, paralizándose importaciones y frenando el ingreso de tecnología para la salud”. Piden, en cambio, cinco años.
Además, se oponen a que todos los dispositivos médicos sean sometidos a control sanitario (aunque el proyecto no propone eso). Y piden que “aquellos dispositivos médicos que ya se encuentran con aprobación de entidades certificadoras de primer nivel mundial, tales como FDA y CE, se les exija un registro documental y se aplique un arancel menor”. Pero la investigación de ICIJ muestra que esas certificaciones son insuficientes.
En la carta a Castro, APIS propone retirar los artículos relativos a los dispositivos médicos para trabajar en una legislación aparte. Un argumento que la industria ha utilizado en el pasado en otros países.
“No hay ningún problema de que la regulación de dispositivos vaya dentro del marco legal de fármacos, ya que ahí sólo se acoge una parte, mientras que la regulación será independiente, con estándares internacionales y enfocada a dispositivos”, dijo Janepsy Díaz, del ISP, a La Tercera.
Han pasado dos décadas desde que la ley definió qué son los dispositivos médicos. Dos décadas en que se dictaron sólo dos decretos para someter a control sanitario apenas cinco dispositivos. Dos décadas en que la falta de regulación ha puesto en riesgo a los pacientes. ¿Cuánto más hay que esperar?
* Este proyecto cuenta con el apoyo del fondo Howard G. Buffet para mujeres periodistas de la International Women’s Media Foundation.




